Proliferative Survival Protocol
This protocol determines the number of proliferating and non-proliferating cells in cell cultures. The protocol involves exposure of cells to 5-bromodeoxyuridine, harvesting them, staining with a buffer that contains Hoechst 33258 dye and a known number of chicken erythrocyte nuclei (CEN). Incorporation of BrdU into the DNA leads to quenching of Hoechst fluorescence. In this way, cells that have undergone one or two rounds of DNA replication can be distinguished from resting cells. To resolve cells in the G1, S and G2 phase of the cell cycle samples are counterstained with ethidium bromide. By dividing the number of cells in the G1, the S and the G2 compartments of the first, second and third cell cycle by the number of CEN their numbers per unit of sample volume is determined.
-
Make the following stock solutions:
- 10 mM 5-bromodeoxyuridine (BrdU; Sigma B-5002) in phosphate buffered saline (PBS). You may need to warm the PBS to 37°C, since 10 mM is close to the maximum solubility of BrdU. Filter sterilize and store at 4°C in the dark. This solution keeps for at least one month.
- Hoechst buffer (see next page)
- 200 mg/ml ethidium bromide. Dilute 200 mL of a 10 mg/ml stock (Sigma: E-1510) into 10 mL distilled water. Store at 4°C in the dark. This solution keeps for at least one month.
- Culture cells for at least one cell cycle duration in the presence of 100 mM BrdU. (See BrdU/Ho/EB protocol in this lab manual for BrdU treatment) Note: some cell types may be sensitive to BrdU, which leads to G2 phase arrest. In case an elevated level of G2 arrest is suspected, it is recommended to test a series of BrdU concentrations (see Rabinovitch et al., 1988). During BrdU labeling, all cell cultures should be protected from light by wrapping all plates or flasks in aluminum foil.
- Harvest cells according to standard procedures (keep cells protected from light all stages of handling).
- Strongly vortex chicken erythrocyte nuclei (named CEN Singlet Cytometry Control; Riese Enterprises, Inc. Grass Vly, CA) and add 1 mL of CEN per mL of Hoechst buffer.
- Resuspend cells in Hoechst buffer; preferably at a density of 0.2 to 0.5 million per mL.
- After at least 15 minutes of staining at room temperature in the dark (usually in a cabinet) add 20 mL of ethidium bromide per mL of cell suspension.
- Stain for another 15 minutes in the dark.
- Analyze samples on the flow cytometer by using the Z-Prol-Surv protocol.
| Final Concentration | Amount to add |
|---|---|
| 0.1 M Tris, pH 7.4 | 50 ml of 1 M Tris pH 7.4 |
| 0.1% NONIDENT P-40 (IGEPAL CA-60) |
500 ml of NONIDENT P-40 |
| 1 mM CaCl2 | 1 ml of 500 mM CaCl2 |
| 5 mM MgCl2 | 5 ml of 500 mM MgCl2 |
| 0.2% BSA | 1 g BSA |
| 2.5 mg/ml Hoechst 33258 | 2 ml of 590 mg/ml Hoechst 33258 |
This protocol determines the number of proliferating and non-proliferating cells in cell cultures. The protocol involves exposure of cells to 5-bromodeoxyuridine, harvesting them, staining with a buffer that contains Hoechst 33258 dye and a known number of chicken erythrocyte nuclei (CEN). Incorporation of BrdU into the DNA leads to quenching of Hoechst fluorescence. In this way, cells that have undergone one or two rounds of DNA replication can be distinguished from resting cells. To resolve cells in the G1, S and G2 phase of the cell cycle samples are counterstained with ethidium bromide. By dividing the number of cells in the G1, the S and the G2 compartments of the first, second and third cell cycle by the number of CEN their numbers per unit of sample volume is determined.
- Open your data files with MPLUS (in case of difficulties with this step ask Mike Shen for assistance).
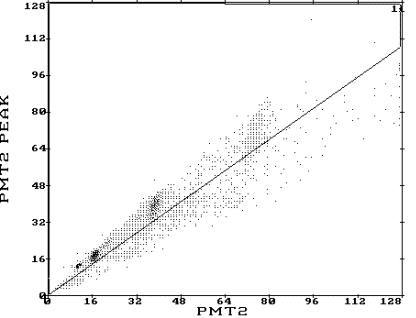
- Click on GATE 2D (blue field on the bottom of the base page).
- Select PMT2 and PMT PK (red options on the GATE 2D page). This will select the PMT2 data (in this assay intensities of Hoechst 33258 fluorescence signals) on the X-axis and the PMT2 PK (peak height of the Hoechst 33258 fluorescence signals) on the Y-axis. A picture similar to the one shown in the top panel of the figure should appear.
- Draw a diagonal as shown in the figure. Single cells will appear above the diagonal, while clumps will appear below the diagonal. This is because clumps have a relatively lower peak height than do single particles of the same total fluorescence intensity.
- Click DONE.
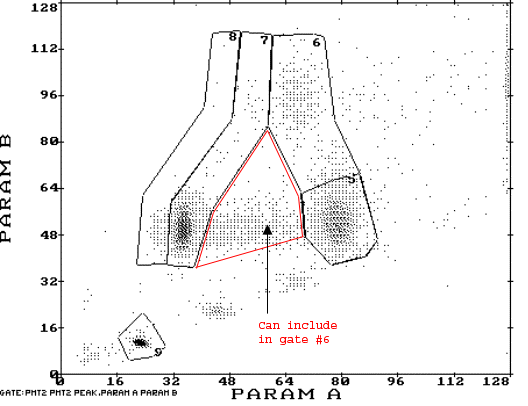
- Select PARAM A and PARAM B. This selects the distribution of the Hoechst fluorescence (quenched DNA) signals of gated cells on the X-axis and the distribution of the EB fluorescence (unquenched DNA) signals of gated cells on the Y-axis.
- Click 'Set Region' to draw gates of different cell cycle phases in various regions (see figure next page)
- G0 cells in region #5
- First cycle (G0 + S + G2) cells in region #6
- Second cycle (G1 + S + G2) cells in region #7
- Third cycle (G1 + S + G2) cells in region #8
- CEN cells in region #9
- Regions from #5 to #9 are set in counter clockwise direction starting from lower right corner for region #5.
- Draw the regions as indicated in the bottom panel of the figure and save the result by clicking on WINPRINT. This saves the image and the signal numbers within each frame.
- After you are done with all your data files, you can exit MPLUS by clicking 'exit' and use PRINT ALL to print all saved files.
There is probably an easier way to describe this, but this is the way I understand.
- List the gates in the following order then enter the number of events in each gate.
- Correct for the number of cells relative to CEN then multiply each corrected number by 10,000
Corrected # cells in G0/G1 Corrected # cells in 1st Cycle Corrected # cells in 2st Cycle Corrected # cells in 3st Cycle (# cells #5 / # CEN) * 10,000 (# cells #6 / # CEN) * 10,000 (# cells #7 / # CEN) * 10,000 (# cells #8 / # CEN) * 10,000 - Calculate the total number of proliferating cell
Add the (Corrected # of cells in the 1st cell cycle) + (Corrected # of cells in the 2nd cell cycle) divided by 2 + (Corrected # of cells in 3rd cell cycle) divided by 4. This gives you the total # of proliferation cells (corrected for the number of divisions they have undergone). Then average the replicates. - Normalize the # of proliferating cells from each treatment group to the control. The control will always be 100%. For example:
| G0/G1 Gate #5 Enter # of cells |
1st Cycle Gate #6 Enter # of cells |
2st Cycle Gate #7 Enter # of cells |
3st Cycle Gate #8 Enter # of cells |
CEN Gate #10 Enter # of cells |
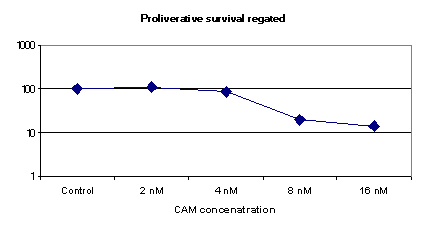
| Group | Total number of Proliferating Cells |
Proliferative Survival |
|---|---|---|
| Control | 1276 | (1276/1276)*100=100% |
| 2nM CAM | 1395 | (1276/1276)*100=100% |
| 4nM CAM | 1024 | (1276/1276)*100=100% |
| 8nM CAM | 259 | (259/1276)*100=100% |